Advantages
- Detection and quantification of over 8,500 proteins in 10 to 100s of fluid and tissue samples.
- TMT based proteomics can reliably detect 30% changes in protein abundance in samples compared to 300% to 400% changes using label free proteomics.
- Compatibility with a wide variety of sample matrices, including tissue, plasma, serum, cell lysates, CSF, exosomes, etc.
- Available for human, rodent, monkey, pig and other higher animal species.
- By using an upfront high abundance protein depletion stage, we can quantify significantly more lower abundant proteins.
- Sample multiplexing in a single TMT based mass spectrometry run significantly increases the sensitivity of low abundance protein detection.
- Ability to enrich for specific protein subsets, such as secreted proteins, phosphopeptides and other post translational modifications and membrane proteins.
- Due to the unbiased nature of the analysis, we report all proteins that are detectable, unlike antibody or aptamer/affimer based technologies where the data is biased to the make-up of the selected multiplexed panel.
- Includes full computational proteomic analysis and bioinformatics covering extensive data interpretation, pathway analysis and biological relevance.
Proteome Sciences is the home of TMT technologies which were invented by one of our employees and we still manufacture all reagents that are sold under license by Thermo Scientific. The introduction of TMT and TMTpro allowed us to multiplex samples early during analysis thereby reducing technical variation and increase the number and quality of protein biomarkers identified. With the latest additions to the TMTpro range we now go up to 18! We also invented LC-MS3 quantification to further improve quantitative accuracy of multiplex experiments offering the most accurate and precise methods for unbiased proteomics.
TMT and TMTpro unbiased discovery is applicable to all biological samples (cells, tissues, fluids) and species. The TMT LC-MS2 workflow is designed for analysis of large cohorts of samples where breadth of coverage is most important. Using the TMT 11-plex, TMTpro 18-plex and now the new TMTpro 35-plex reagents we can analyse 10/17/34 samples per experiment with the 11th/18/35th channel filled by a pooled whole-study reference. This allows normalisation of MS data across multiple 11-plex to 35-plex sets and provides highly sensitive detection of changes in protein expression in cohorts of 10s to 100s of samples. The typical TMT LC-MS2 workflow is shown in Figure 1.
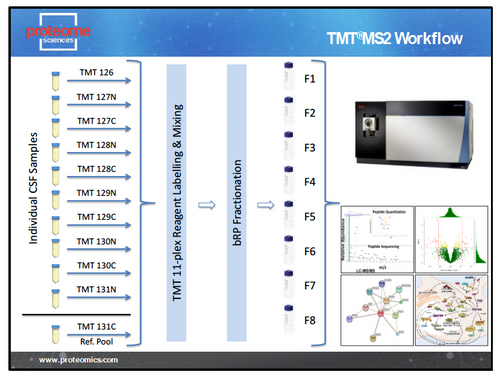
Figure 1 – TMT LC-MS2 Workflow.
For each set of up to 11, 18 or 35 plex samples we generate a single 11, 18 or 35 TMT labelled sample that is fractionated off-line and each of the 8 resulting fractions analysed for 3h on an Orbitrap Tribrid mass spectrometer (Thermo Scientific). Peptide and protein identification and TMT reporter ion intensities are obtained from Proteome Discoverer (Thermo Scientific). Data is then processed through our standard bioinformatics pipeline.
For studies where greater quantitative accuracy is required, we can use LC-MS3 level quantification using the synchronous precursor selection (SPS) workflow. This does not affect the study design or timelines, but does reduce protein identification rates by around 5% in a typical study.
The same principles if quantification are employed in our other main workflows such as Super Depletion, TMTcalibrator and SysQuant.
An example report is available upon request.
Sample requirements for LC-MS2 or LC-MS3 analysis (per 18-plex experiment).
| Application |
Plasma/Serum (per sample)
|
CSF (per sample) | Tissue Trigger (total protein) |
|---|---|---|---|
| Standard LC-MS2 | 100 ul | 250 ul | 1.3 mg |
| LC-MS2 with upfront Super Depletion | 250 ul | Not applicable | 1.3 mg |
| SysQuant phosphoproteomics | Generally not applicable | 600ul | 7.4 mg |
For FAQ’s on Unbiased Protein Biomarker Discovery based Proteomics please follow this link.
| What’s included |
|
| Material required |
|
| Typical turnaround time |
|